Mixed Ligand Monolayer Self-Assembly on Nanoparticle Surfaces
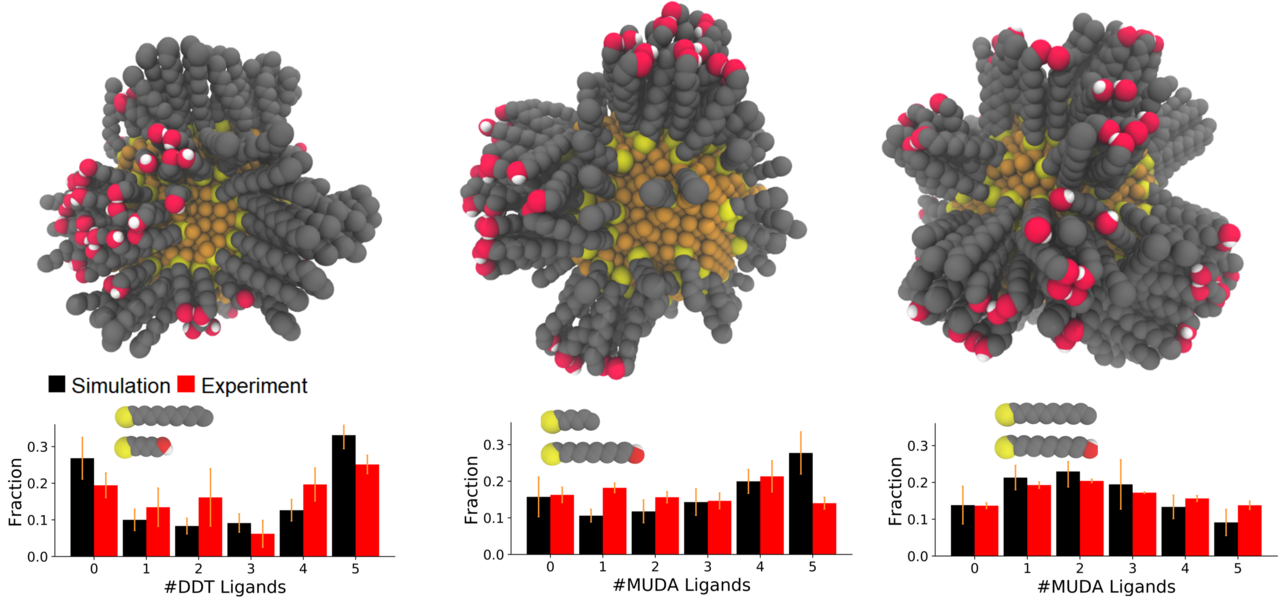
Nanoparticles (NPs) protected with self-assembled ligand monolayers (SAMs) have applications ranging from photonics and catalysis to drug delivery and biosensing. SAMs with a mixture of ligands may self-organize into random, striped, patchy, and Janus-like surface morphologies. These patterns impact their functional properties but are difficult to predict. Even the characterization of SAM morphology on NPs <10 nm has proven difficult. In an NSF-funded collaboration with Prof. David Green at UVA, who employs matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) to indirectly characterize NP-SAMs, we adapted an advanced Monte Carlo (MC) technique developed by Siepmann and Frankel to equilibrate atomistic models of these sluggish mixed ligand monolayers – monolayers which take days to equilibrate in the lab.
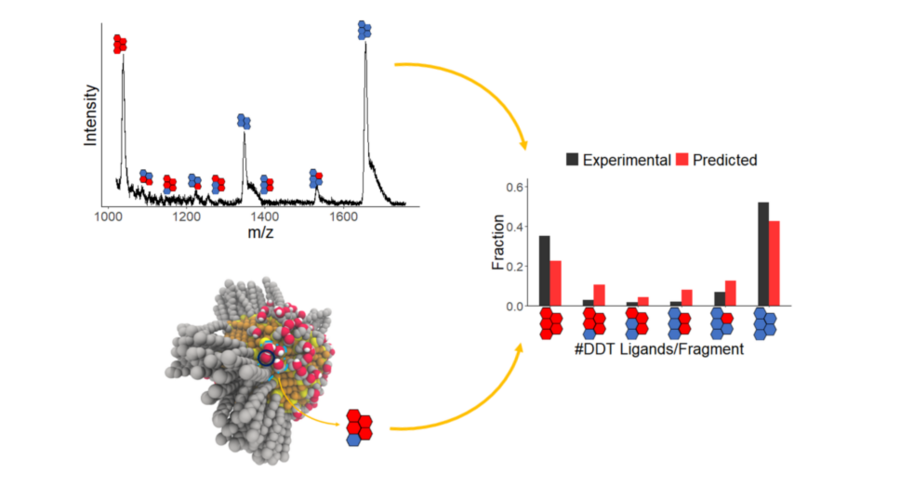
Key Findings: The Green group synthesized silver NPs with SAMs consisting of dodecanethiol (DDT) and mercaptoethanol (ME) at varying ratios and analyzed them using MALDI-MS. The results suggested that DDT and ME phase separated on the NP surface. We then employed atomistic MC simulations to generate equilibrated SAM morphologies. To quantitatively compare the computational and experimental results, we developed a method to calculate expected MALDI-MS spectra from the resulting atomistic models. The simulations agreed quantitatively with the experiments, both indicating that the DDT/ME ligands undergo phase separation in the SAMs, resulting in large, patchy Janus-like domains (Merz, ACS Nano, 2018). We then applied the same MC approach to successfully predict MALDI-MS results for three new ligand combinations, which were chosen to test the ability to separately tune SAM morphology and chemical functionality. The resulting morphologies ranged from randomly-mixed to Janus-like, demonstrating that chain-length modifications are an effective way to tune SAM morphology without needing to alter chemical functionality. Our results also provided atomistic detail of the SAM structures, such as patch sizes and co-crystallization patterns (Merz, Soft Matter, 2019).