Sequence Formation of Self-Assembling Nascent Copolymers
Advanced copolymers – polymers made up of more than one type of monomer unit – have the potential to revolutionize important material applications, such as protective coatings for everything from ships to surgical implants, nanoscale delivery vehicles that release drugs precisely where they are needed, and water filtration membranes to improve global health. The functional properties of copolymers depend on the sequence in which their monomer units are bound together. Unfortunately, sequences of synthetic copolymers remain difficult to control, hindering the design and development of much needed materials.
Using coarse-grained simulations, my group is building a comprehensive understanding of the forces that govern sequence in step-growth reactions. Our results revealed for the first time how the emergent self-assembly of nascent, growing oligomers can influence sequence in even simple, one-pot step-growth polymerizations. This work is supported by an NSF CAREER Award and has won recognition through a Junior Faculty Prize at the Berkeley Stat Mech Meeting and an ACS OpenEye Outstanding Junior Faculty Award.
Key Findings: Through our simulations, we discovered that relatively weak attractions among nascent oligomers can exert considerable influence over the final sequences of generic A,B-copolymers. The effects we observe cannot be accounted for within conventional polymerization theories due to a breakdown in the applicability of Flory’s principle of equal reactivity. However, we found they could be readily explained by the physics underlying Flory-Huggins theory, since the simulations show nascent chains assembling as they lengthen. Microphase separations between A-rich and B-rich regions thus emerge from the copolymerization process itself and bias subsequent bond formation (Zhang, Macromolecules, 2019). The copolymer’s flexibility modulates this effect. We observed that nascent chains align within the polymerization-induced aggregates as chain stiffness increases, which influences the resulting sequence. For persistence lengths ≥10 monomers, modest effective attractions between monomers of the same type yield relatively uniform homopolymer sequence blocks. Our results show that, for copolymer systems with physically reasonable interactions and flexibilities, a copolymer’s persistence length can influence its own sequence (Zhang, J Phys Chem B, 2021). In recent work, we discovered that the characteristic length of these homopolymer blocks is determined by the ratio between the reaction and diffusion timescales (manuscript in preparation), providing a new route towards block length control through the tuning of solvent viscosity. Our results also show that a solvent selective for one type of monomer also influences sequence and yields hierarchically ordered core-shell-like structures (Hamblin, Soft Matter, 2022). Finally, by applying a more formal analysis of the ideal reaction kinetics and Mayo-Lewis ratios to investigate variations in both activation energies and non-bonded attractions, we quantified the impact on sequence that arises solely from emergent microphase separations and observed how activation energies and non-bonded attractions can work in concert or in opposition while biasing sequence (Nguyen, ChemRxiv, 2022).
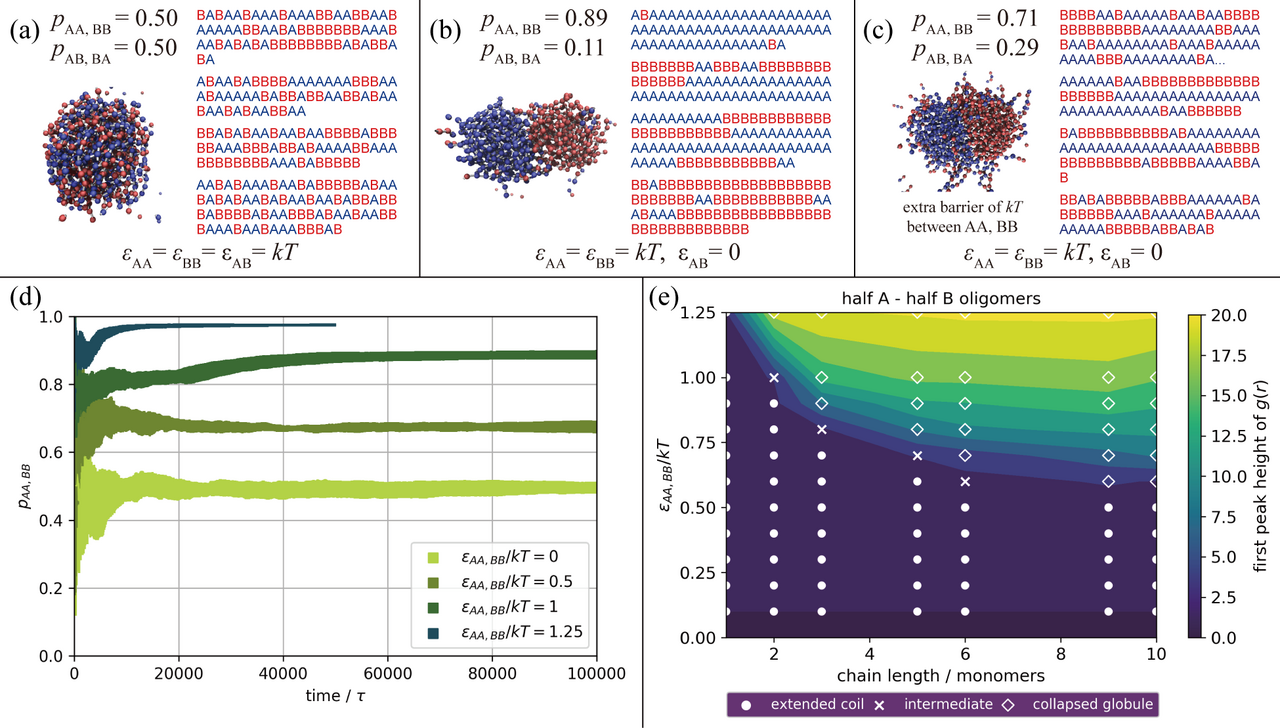
Figure 2: Simulation Results of a Step-Growth A,B Copolymerization. We show here our results from simulations using the model above for an A (blue) and B (red) copolymer. A simple Lennard-Jones-like interaction (where repulsions are held fixed) exists between the type 1 particles within each bead, and the \(\epsilon_{AA}\), \(\epsilon_{BB}\), and \(\epsilon_{AB}\) values indicate the well-depth of the attractive portion of that non-bonded potential for each monomer combination. For (a), (b), and (c), snapshots are shown of the system after polymerization alongside the sequences of a sample of the resulting oligomers. The probabilities of neighboring monomers along the chain having the same identity, \(p_{AA,BB}\), or the opposite identity, \(p_{AB,BA}\), are also reported. In (a), there are equal attractions between all monomers, like and unlike; in (b), there are only attractions between like monomers; and in (c) there are only attractions between like monomers, but there is also an additional barrier of \(k_BT\) for the reactions between like monomers. Steric repulsions and reaction barriers are otherwise fixed to be the same between all pairs. In (d), \(p_{AA,BB}\) is plotted vs. the reaction time for a series of simulations with different strengths of non-bonded attractions between like monomers (here \(\epsilon_{AB}\) = 0). Finally, in (e), the phase behavior of non-reactive homo-oligomers of varying lengths (with half of the oligomers containing A monomers, and half containing B), with like attractions of varying strengths (again \(\epsilon_{AB}\) = 0), is explored through the use of a visual assessment of chain structure (symbols) as well as the height of the first peak in the corresponding radial distribution functions for monomers not directly bound to one another (color-scale). See Zhang, Macromolecules, 2019.