Research
Overview
Dearomatization of Aromatic Molecules Using Transition Metal Complexes
Aromatic molecules—benzene, pyridine, furan, and their relatives—are among the most abundant and inexpensive starting materials in chemistry, but they present a fundamental problem for drug discovery: they are flat. The remarkable stability of aromatic rings, arising from their delocalized electrons, means that converting them into three-dimensional, functionalized structures using conventional chemistry is difficult and limited in scope. Yet pharmaceutical researchers increasingly recognize that molecular shape—particularly three-dimensionality and the presence of multiple stereocenters—is critical to how a drug candidate behaves in the body. The Harman Research Group addresses this challenge by developing a fundamentally new way to break the aromaticity of these rings and build complex, three-dimensional molecules from them.
The central strategy exploits the ability of certain electron-rich transition metal complexes—specifically those based on tungsten and molybdenum—to bind to just two adjacent carbons of an aromatic ring, a mode of attachment called dihapto (η²) coordination. Unlike classical approaches in which a metal binds all six carbons of a benzene ring and activates it toward substitution reactions, these dihapto-coordinated complexes disrupt aromaticity while leaving most of the ring exposed and highly reactive toward addition of carbon-based electrophiles and nucleophiles. In essence, the metal transforms a chemically inert aromatic ring into a reactive diene. The tungsten complex {WTp(NO)(PMe₃)} and its molybdenum analogue {MoTp(NO)(DMAP)} are the workhorses of the program; they are stable, can be prepared on gram scales, and bind a remarkably wide range of aromatic substrates—including benzenes, naphthalenes, phenols, anilines, pyridines, pyrroles, furans, and thiophenes.
Classically, dearomatization is effected by the binding of an arene to a first-row transition metal, giving complexes such as Cr(CO)3(η6-arene) and Mn(CO)3(η6-arene)+. The bound arenes become susceptible to nucleophilic addition, ultimately yielding substituted arenes or dienes derived thereof, and the metal is typically discarded after this single transformation. The history of this type of dearomatization spans over half a century and is well-established in modern chemistry.
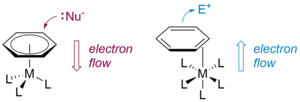
The Harman research group has developed an alternative and complementary approach to dearomatization, in which the arene is bound to the metal (Os, Re, Mo, or W) in a dihapto (η2) manner. Here, the metal-arene bond is primarily stabilized by the interaction of a filled dπ orbital of the metal with an unfilled π* orbital of the arene. This "π back-bonding" activates the bound arene towards electrophilic addition, rather than nucleophilic addition. Furthermore, the increased electron density partially dearomatizes the η2-bound ligand, thus allowing bonds to be formed at the non-coordinated carbons of the π system.
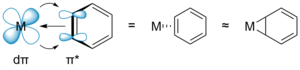
Once an aromatic molecule is bound to the metal, a rich and controllable chemistry unfolds. Sequential addition of electrophiles and nucleophiles to the activated ring allows the construction of highly substituted cyclohexenes, polycyclic frameworks, and heterocyclic scaffolds that are difficult or impossible to make any other way. Importantly, the metal also acts as a stereochemical director: because new bonds form on the face of the ring opposite the metal, multiple new stereocenters are set simultaneously and with high selectivity, often exceeding 20:1 in diastereomeric ratio. When a resolved (single-handed) metal complex is used, this stereocontrol extends to enantioselectivity, providing access to enantioenriched products. The molybdenum system offers additional practical advantages—lower cost and the ability to recycle the metal fragment after the organic product is released—making large-scale synthesis feasible.
The broader significance of this work lies in what organic chemists call "escaping flatland." Most molecular libraries used in drug discovery are dominated by flat, aromatic molecules, partly because the reactions available to make them are well established. By providing a practical toolkit to convert simple aromatic precursors into three-dimensional structures with multiple stereocenters and diverse ring systems, our dearomatization methodology opens chemical space that is otherwise inaccessible. Future directions include extending the approach to catalytic and enantioselective molybdenum systems, exploiting redox-catalyzed reaction manifolds, and applying these methods directly in medicinal chemistry programs targeting novel molecular architectures. Just as a Lewis acid can modify the reactivity of any molecule bearing a lone pair, these metal complexes have the potential to alter the chemical behavior of virtually any molecule containing a π bond—a capability with far-reaching implications for the synthesis of biologically active compounds.