3.1. Solvated Adsorbate Diffusivities Depend on Surface Curvature
Inspired by previous work showing that variations in surface curvature and CNT helicity can alter the diffusional pathways of an atomic adatom on a CNT surface [
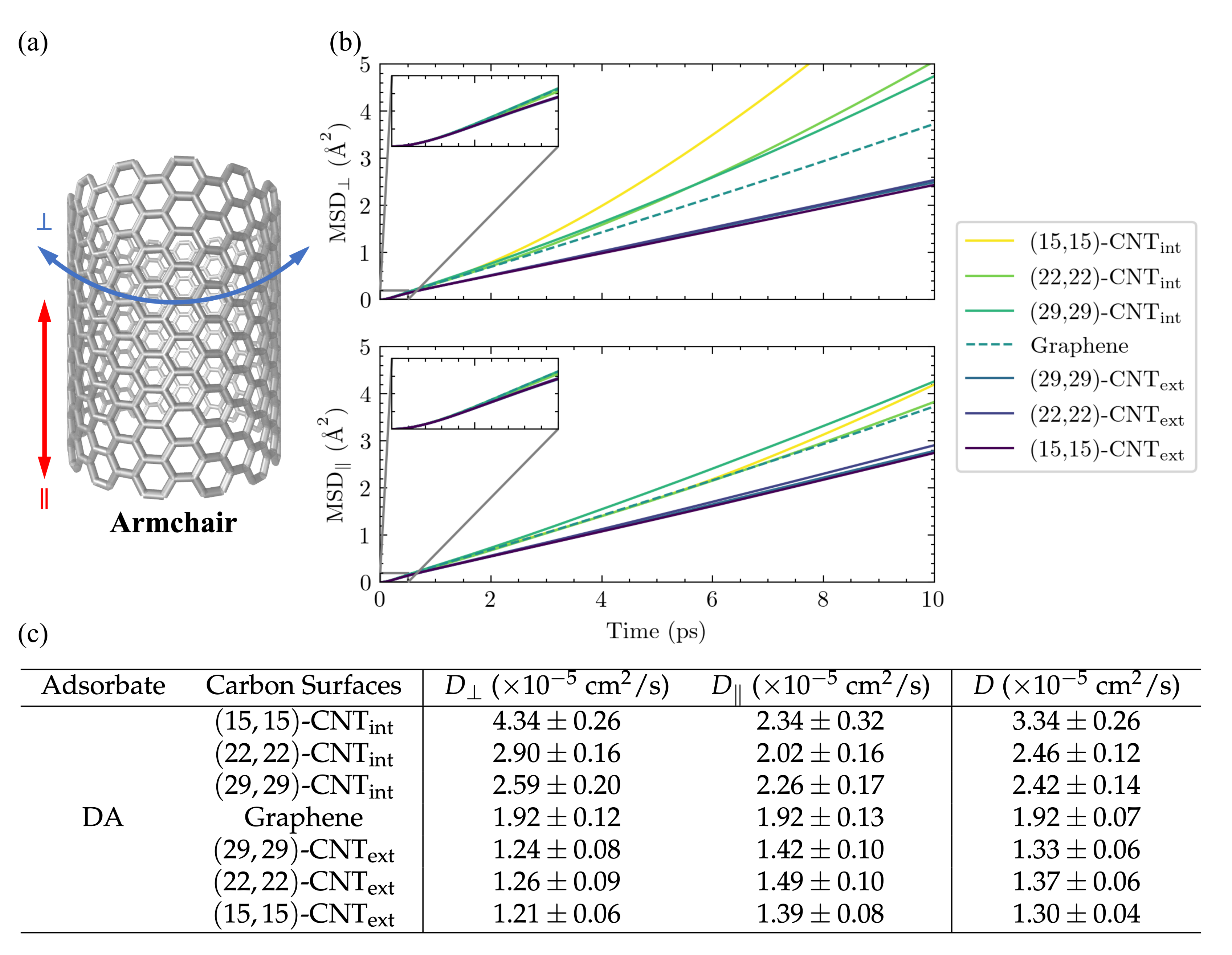
23], we set out to investigate the motion of DA across a series of solvated CNT surfaces with varying curvatures. The mean squared displacements (MSDs) of the adsorbates along the CNT axis (MSD
) and around its circumference (MSD
) are plotted in
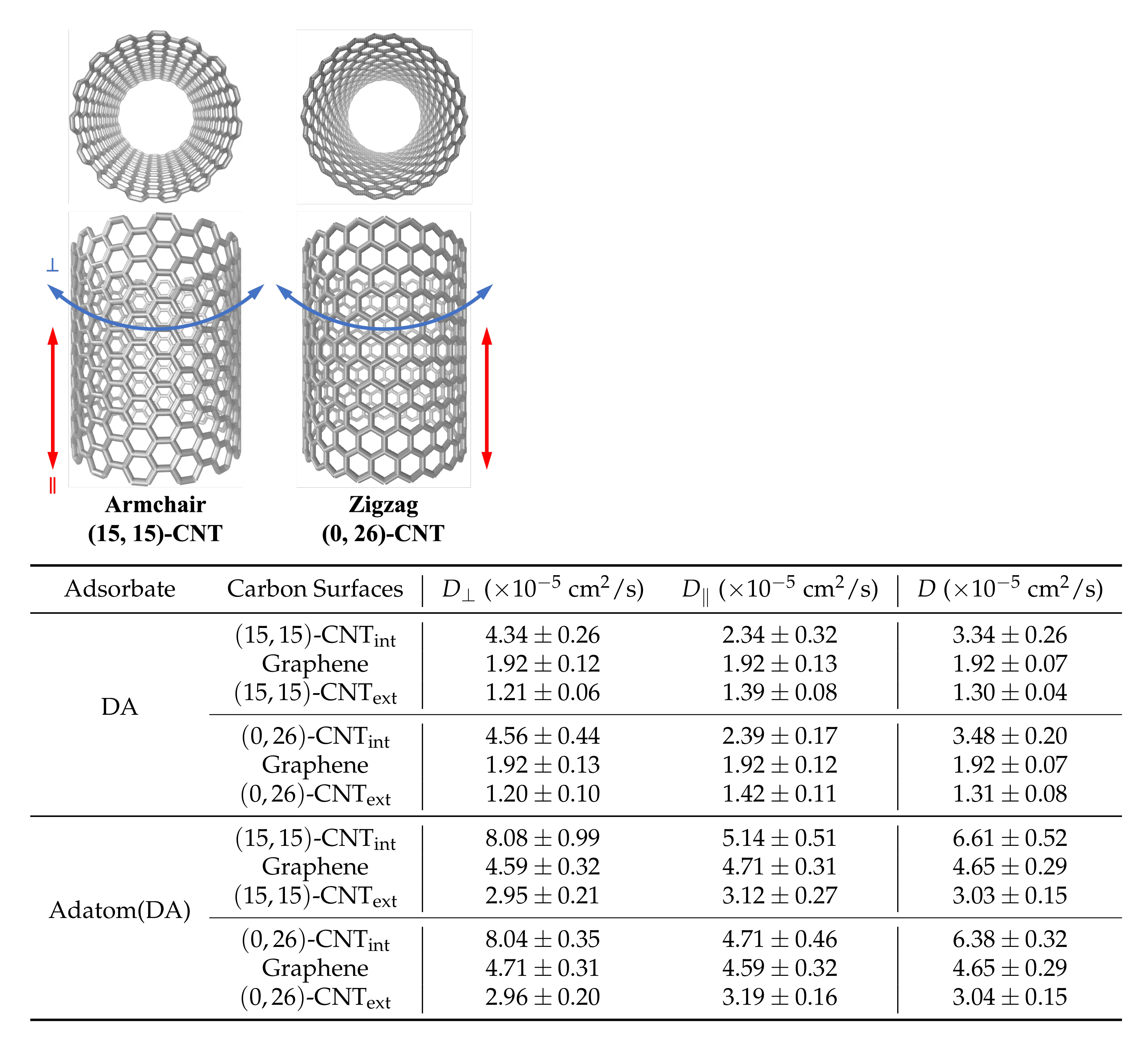
Figure 3b as a function of time for DA on seven differently curved armchair CNT surfaces: three on the CNT interior, one on flat graphene, and three on the CNT exterior. The 1D diffusivities,
and
, and the overall 2D diffusivities,
D, are listed in
Figure 3c. The diffusion coefficients,
D, were computed from the MSDs using the Einstein relation, [
26,
44] as detailed in the
Supplementary Materials.
The observed diffusion constants are smallest on the convex CNT exterior and largest on the concave CNT interior. The largest shifts with curvature are seen in the values, in keeping with the direction in which the surface curves. In addition, a clear increase is seen in the values among the CNT interior results as the concavity increases from -CNT to -CNT.
In order to compare the resulting diffusion constants to experimental values, we adjust them to correct for an unphysical system size dependence. This known finite-size effect [
37,
38,
39,
46] is discussed in more detail in the
Supplementary Materials (see
Figure S4 and accompanying text), and the values of the diffusion constants extrapolated to the infinite system size,
, are shown for a subset of the cases in
Table 1. In our simulations, the extrapolated 2D diffusion coefficient of DA on flat graphene is
, while its value ranges from (1.1–2.5)
for DA on differently curved CNTs. For comparison, the 3D diffusion coefficient calculated for DA from flow injection experiments is
[
47].
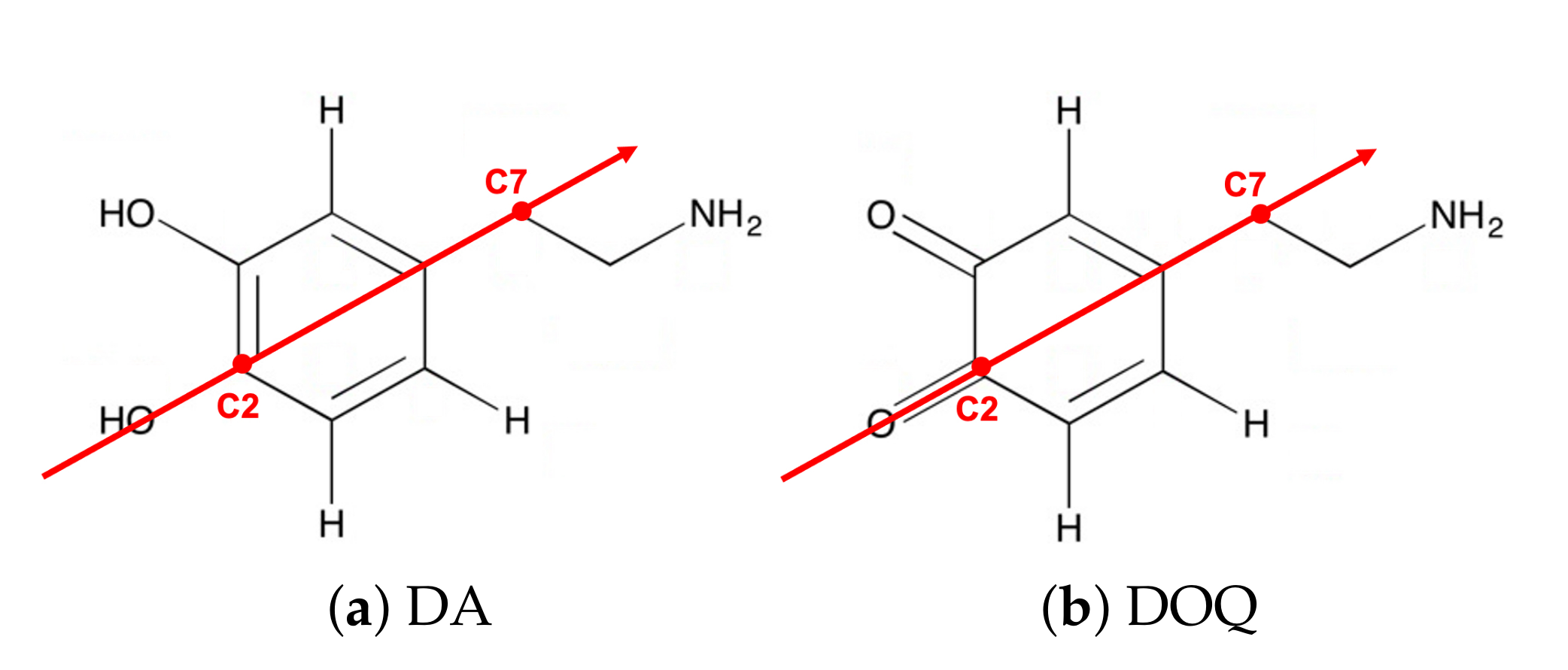
Curvature dependence is observed for DA, DAH, DOQ, and DOQH.Table 2 presents the diffusion constants obtained for these four species on both the interior and exterior surfaces of
-CNT, along with the flat graphene results. From these measurements, we find that the curvature-dependence is similar across all four species.
In addition, across all three curvatures we find that the protonated species, DAH
and DOQH
, diffuse more slowly than their neutral counterparts, DAH and DOQ, while the oxidized species, DOQ and DOQH
, diffuse more rapidly than their reduced counterparts, DA and DAH
. These trends were previously observed on flat graphene [
18] and can be readily explained by differences in the interactions of each species with the solvating water molecules: the positively charged species have increased interactions with the polar solvent, while the oxidized species have reduced interactions with the solvent—their quinone moieities are only able to act as hydrogen bond acceptors, as compared to the reduced diol moieties, which can act as both hydrogen bond donors and acceptors. Increased attractions with the solvent will increase the adsorbate’s effective hydrodynamic radius,
, which is inversely related to the diffusion constant,
D, of a solvated sphere in nonturbulent flow via the Stokes–Einstein equation [
36]:
, where
is the Boltzmann constant,
T is temperature,
is solvent viscosity, and
c is a constant that describes the boundary conditions at the solvent-sphere interface. The Stokes–Einstein equation cannot be rigorously applied here for these partially solvated, small molecular adsorbates; however, it qualitatively explains the observed trends.
Our simulation results show that DA diffusion clearly depends on placement on the inner or outer surface of the CNT, with enhanced motion on the CNT interior. Overall, this observed curvature dependence is consistent with the general trends observed previously for an atomic adatom [
23].
3.2. Dependence Does Not Arise from Curvature-Induced Shifts in Surface Roughness
In the case of the previously studied atomic adatom, the observed reduction of diffusion barriers for the adatom on a surface with negative curvature (the CNT interior) resulted from the smoothing of the carbon energy surface as it changes from convex to flat to concave [
23]. However, it is not clear how this effect functions for a molecular adsorbate such as DA, which is larger than the underlying hexagonal carbon structure, flexible, and asymmetric in shape with an uneven charge distribution. In addition, the role of solvent was not considered in the prior work and may mitigate the influence of surface energy roughness. In this section, we probe the role of surface roughness in this system by investigating how the lateral distributions of these adsorbates depend on curvature, how their diffusivities depend on CNT helicity, and how their diffusivities depend on curvature in the absence of solvent.
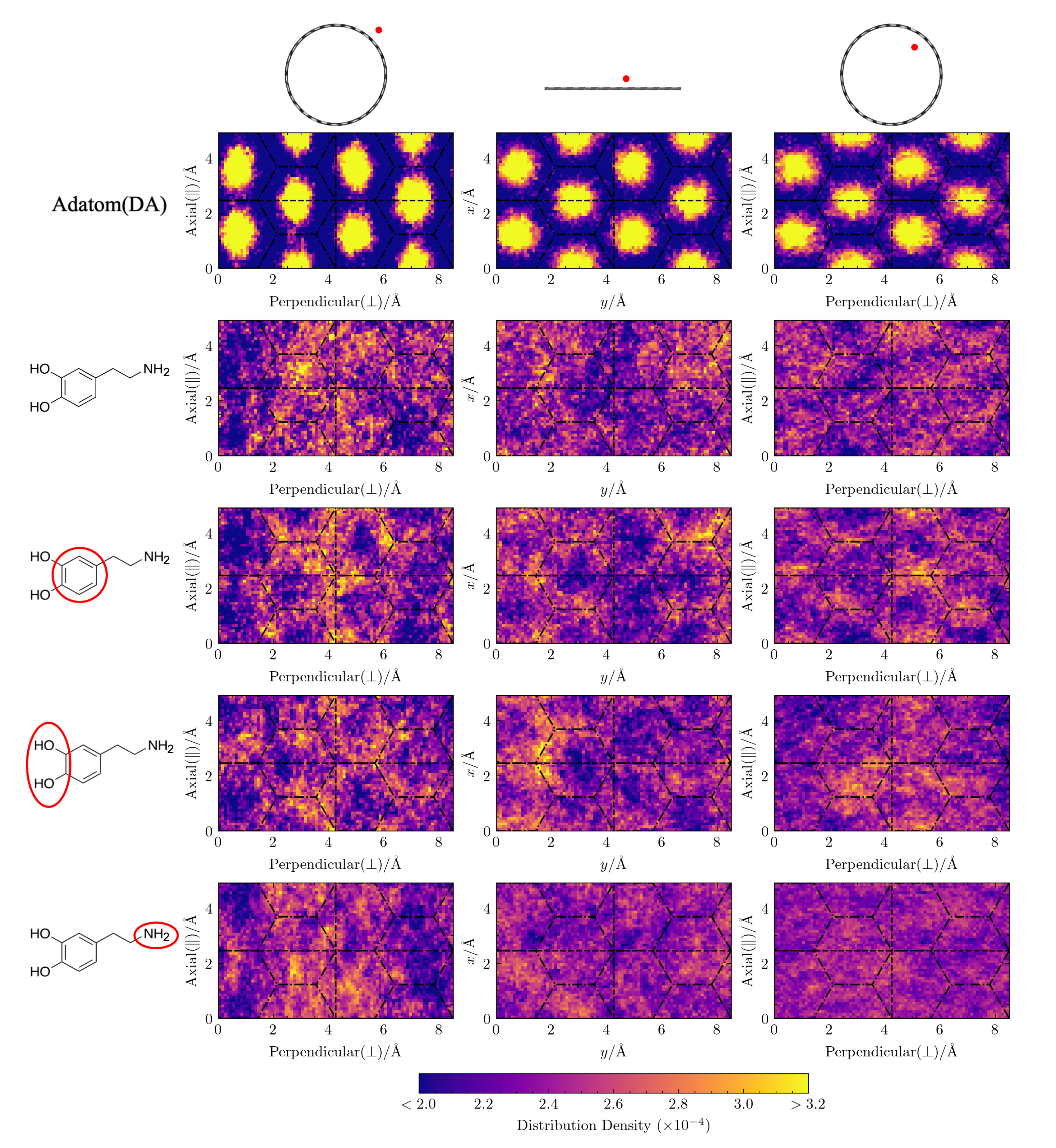
Lateral distributions of molecular adsorbates are similar across curvatures. In
Figure 4, we plot the lateral distributions for the adatom(DA), DA, and its moieties on three differently curved surfaces: the exterior of a
-CNT nanotube, flat graphene, and the interior of a
-CNT nanotube. By comparing these distributions to the underlying hexagonal aromatic ring pattern of the carbon surfaces, we can observe how curvature-induced differences in the energy surface roughness influence the placement of these atomic and molecular adsorbates.
First, we consider the lateral distributions of adatom(DA), an atomic adatom with the same mass as DA. Shown in the first row of
Figure 4, these distributions clearly display the characteristic hexagonal pattern that corresponds to the centers of the honeycomb structure of the aromatic carbon surface. As the surface curvature changes from convex to concave, the lateral distribution of adatom(DA) gradually becomes more uniform, as expected from the previously noted smoothing of the surface energy as the curvature becomes more negative [
23]. Despite the presence of solvating waters in our simulation, the dependence of adatom(DA)’s lateral distribution on the underlying carbon structure and its curvature persists.
In contrast, the lateral distributions of DA’s COM and that of its constituent moieties, as shown in the next four rows of
Figure 4, display almost no dependence on the underlying hexagonal carbon structure and we see no clear trend in the distributions with curvature. This lack of structuring and curvature dependence suggests that the underlying surface energy roughness is not a dominant factor in determining the lateral placement of DA, which extends spatially over a region larger than the hexagonal lattice spacing of the underlying carbon surface.
Diffusion coefficients for zigzag and armchair CNTs are indistinguishable. Helicity-dependent diffusion of atomic adsorbates on CNT surfaces has been previously observed in simulations, where different diffusive pathways were observed on armchair and zigzag CNT surfaces due to the surface energy landscapes that emerged upon curving graphene in different directions [
23,
48]. To probe this effect for our solvated system, we simulated the diffusion of both DA and adatom(DA) on the interior and exterior surfaces of highly curved armchair and zigzag CNTs.
Figure 5 shows the two CNT structures with 10 Å radii (
-CNT and
-CNT) as well as the
,
, and 2D
D values obtained from these simulations. The corresponding results on flat graphene are also shown in each case for comparison.
We found no significant difference between the armchair and zigzag diffusion constants in our simulations for either the atomic or molecular DA adsorbates. This result is expected for DA itself, given the insensitivity of its lateral distribution to the underlying hexagonal structure in
Figure 4. The shift in the lateral distribution for adatom(DA) with curvature, however, suggests that differences between zigzag and armchair diffusivities are possible within our system. Even so, the results for the two cases are statistically indistinguishable, perhaps due to the dominant influence of surface hydration on adsorbate dynamics in these systems which we found in our prior work on flat graphene [
18].
Curvature dependence of D disappears in the absence of solvent. The negligible influence of the carbon surface’s hexagonal patterning on DA’s lateral distributions in
Figure 4 suggests that the differences in
D between the CNT surfaces of various curvature in
Figure 3 and
Table 1 do not actually arise from curvature-mediated changes to the energetic interactions between the adsorbate and the surface. The lack of dependence of DA diffusion on CNT helicity in
Figure 5 supports this conclusion.
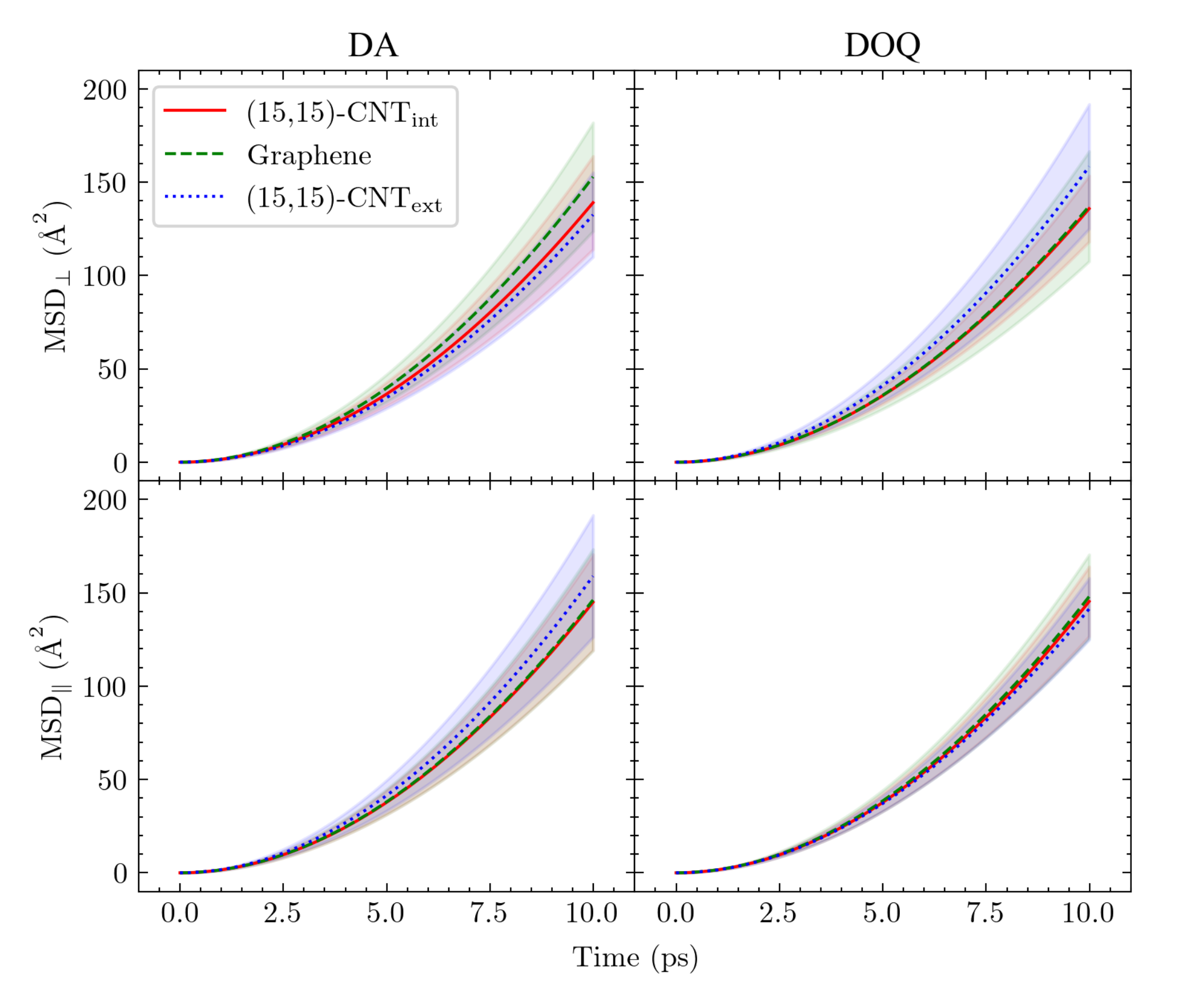
To isolate the adsorbate–surface interactions and further probe this dependence directly, in
Figure 6 we plot MSD
and MSD
for DA and DOQ on the carbon surfaces in the absence of solvent. Only the neutral species are simulated due to the lack of charge balance under vacuum conditions. The average MSD results for the adsorbates on flat graphene and on the interior and exterior of the
-CNT at the vacuum interface are shown plotted with lines, while the noise on each measurement is indicated by the shaded regions.
The resulting curves are not linear over the time regime plotted, indicating that inertial motion lasts for much longer times at the carbon:vacuum interface than at the carbon:water interface. Since the simulations do not allow for carbon surface fluctuations, which would be expected to significantly reduce the timescale of inertial motion decay in the absence of solvent, these MSD curves are only useful as a way to isolate the direct interactions between the adsorbate and the different carbon surface architectures and test their influence on adsorbate diffusion.
The results within each panel show significant overlap of the shaded regions and no observable curvature dependence. In addition, the difference between the diffusivities of DA and DOQ disappears, as expected from our conclusions above regarding the importance of solvent and the effective
in determining the relative diffusivities of DA, DOQ, and their protonated species [
18]. Finally, even the MSD
and MSD
curves appear identical, indicating that the differences observed in CNT surface diffusion between the axial and perpendicular directions in
Figure 3 and
Table 1 are also attributable to solvent effects.
Taken together, these results suggest that the curvature dependence that we observe in the diffusion constants for adsorbed DA and DOQ at the carbon:water surface do not actually arise from curvature-induced changes in the energy surface roughness, as was the case in the prior work on an unsolvated atomic adatom [
23]. Instead, we conclude that the curvature-dependence of these molecular adsorbates’ diffusivities arises from a more complex interplay of surface curvature and surface solvation.
3.3. Adsorbate Structure Depends on Curvature, Charge, and Solvation
In this section, we investigate in detail the adsorbate’s configuration on the surface and its dependence on curvature, charge, and solvation. First, we examine the vertical placement of DA and DOQ and its constituent moieties above the different carbon surfaces; in particular, we examine the various configurations available to the amine group. Then, we consider the tilt angle of the aromatic ring above the surface and the adsorbate’s orientational alignment with the CNT axis. Finally, we consider how the differently curved surfaces shift the number of water molecules in the first solvation shell around the adsorbate, which will influence the effective hydrodynamic radius, , and, therefore, the diffusivity.
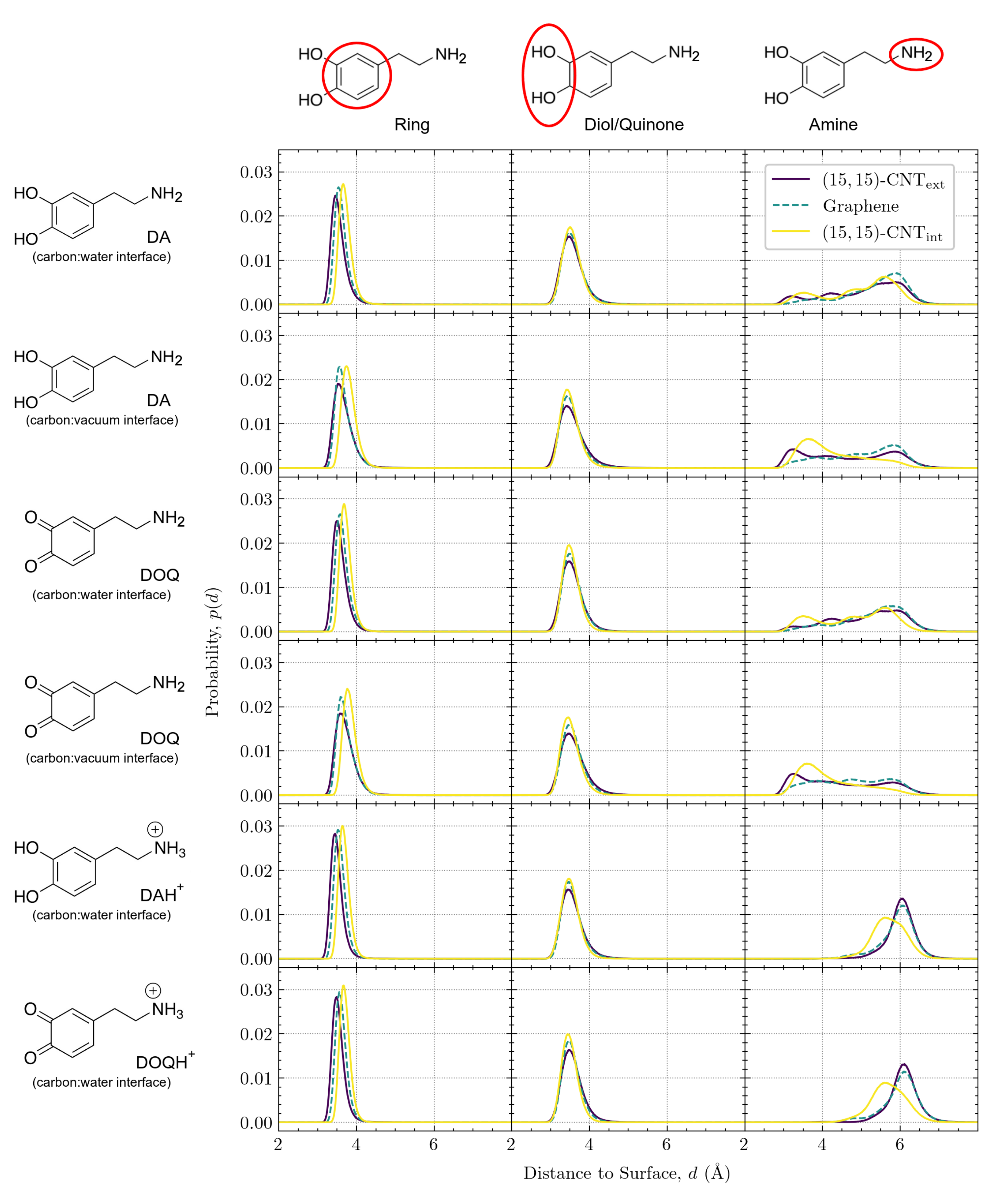
The distance of the adsorbate above the surface depends on curvature, charge, and solvation. The vertical distance,
d, is defined as the distance between the COM of a moiety and its closest point on the carbon surface.
Figure 7 displays the vertical distributions for the aromatic ring (left column), the diol/quinone (middle column), and the amine group (right column) on the three surfaces. DA and DOQ distributions are shown at both the carbon:water and carbon:vacuum interfaces, while DAH
and DOQH
distributions are only shown at the carbon:water interface.
In the left column of
Figure 7, the position of the aromatic ring for all solvated species shifts slightly away from the surface as its curvature changes from convex to flat to concave. Due to the ring’s structural rigidity, its COM can get closer to the surface when adsorbed on the convex exterior of the CNT than when adsorbed to its concave interior, where interactions with the inward-curving walls shift the center of the ring slightly away from its optimal distance on the flat surface. Interestingly, for the two cases of DA and DOQ at the carbon:vacuum surface, the aromatic ring distributions for both the exterior and interior CNT surfaces shift slightly to the right, as compared to the solvated cases. This shift away from the surface indicates the importance of solvation in determining the optimal vertical position for the CNT-adsorbed aromatic rings.
In contrast, the diol/quinone moiety distributions in the middle column of
Figure 7 display no shift with curvature, although the peak narrows slightly in all cases as the curvature of the carbon surface changes from convex to flat to concave. The invariance of these peaks, coupled with their location on the edge of the aromatic ring, provide further evidence that the shift in aromatic ring placement with curvature reflects constraints on the optimal surface ring distance due to the curved surface geometry.
Finally, in the right column of
Figure 7, we plot the vertical distributions of the amine tail, which is tethered to the aromatic ring through rotatable bonds and can thus adopt a variety of configurations. Our prior work on flat graphene [
18] demonstrated that the vertical distribution of the amine group is sensitive to its protonation state, as the positively charged DAH
and DOQH
amines can form additional hydrogen bonds with the bulk phase water molecules. These prior observations showed that the neutral amine vertical distributions have three peaks and span a range of about 3–7 Å from the surface, while the positively charged amines have a narrower distribution around a single peak at ≈6 Å from the surface. Similar overall distributions are seen for the CNT exterior and interior surfaces, with a broad, three-peaked distribution for the neutral species and a narrower distribution further from the surface for the charged amines. However, as the curvature changes from the exterior to flat graphene to the interior, the amine distributions are altered, especially for the CNT interior. In addition, we find that the distributions shift closer to the surface for both DA and DOQ at the carbon:vacuum interface as compared to their corresponding distributions at the carbon:water interface.
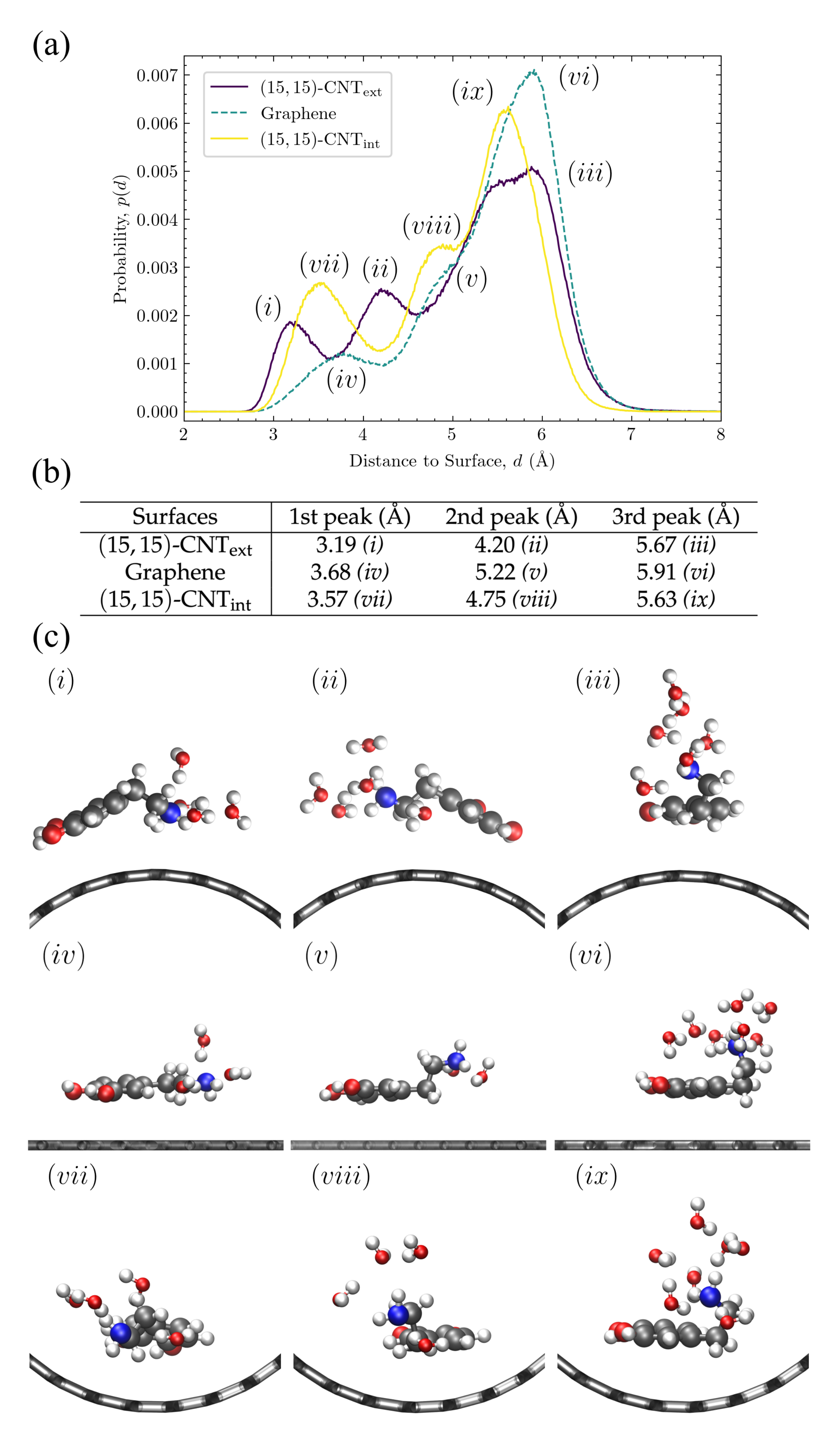
Amine configurations are highly variable and display significant curvature dependence. Given the complex variation observed in the amine vertical distributions, in
Figure 8, we investigate in more detail these distributions for DA at the carbon:water interface. The three peaks for the CNT
, flat graphene, and CNT
distributions have been marked with letters in
Figure 8a, their positions are listed in the table shown in
Figure 8b, and a sample configuration at the characteristic distance within each peak is shown in
Figure 8c.
The peaks closest to the surface in
Figure 8a
(i, iv, vii) correspond to configurations in which the amine group is in close contact with the surface. These first amine distribution peaks are observed at 3.1–3.7 Å (see
Figure 8b) on all three surfaces, which is close to the sum of the van der Waals (vdW) radii, 3.27 Å, of a carbon with a nitrogen in the OPLS-AA force field [
33]. The amine groups in these configurations are closest to the water molecules in the first layer near the surface, as can be seen in the corresponding sample structures in
Figure 8c. The first and the second peaks in the density profile of water are observed at ≈3.3 Å and ≈6.2 Å, respectively (see
Figure S5). The second set of peaks in the amine distribution
(ii, v, viii) are observed at 4.2–5.2 Å. The amine groups in these configurations are therefore likely to form hydrogen bonds with both the first and second layers of water molecules, as shown in the sample structures in
Figure 8c. Last, the third set of peaks
(iii, vi, ix) are seen at 5.6–5.9 Å, which is closest to the water molecules in the second layer. The sample configurations for these peaks in
Figure 8c show the amine tail stretching up toward the bulk water.
Although the presence of these three peaks persist across the curvatures, their locations shift with curvature, as can be seen in
Figure 8a. When the curvature changes from convex (purple) to flat (teal), all three peaks shift rightwards. When the curvature changes from flat to concave (yellow), these three peaks shift back toward the left but to a lesser degree.
To understand the trend in the peak closest to the surface, we consider the three structures shown on the left in
Figure 8c. “Tentlike” configurations similar to
(i) are more likely on the convex surface, where the amine group reaches down toward the carbon surface. Even though the center of the aromatic ring is slightly tilted away from the surface, it remains closer to the surface than it would in a similar configuration on a flat or concave surface. The position of the amine as it points down toward the surface corresponds to the leftmost peak in
Figure 8a, at 3.19 Å. The structures
(iv) and
(vii) also contain amines quite close to the surface, but given the mismatch between the surface curvature and the tentlike structures of
(i), they are not able to get as close, showing a peak distance of 3.68 Å on the flat graphene surface and of 3.57 Å on the convex surface (see
Figure 8b).
For the middle peaks,
, represented by the corresponding structures in the middle column of
Figure 8c, the leftward shift is even stronger for DA on the convex surface
(ii) and represents another version of the “tentlike” structures—one with the same tilted aromatic ring but with the amine group pointing back toward the solvent as in structure
in
Figure 8c. In the next section, we discuss the distributions of these aromatic tilt angles and their curvature dependence. On the flat and concave surfaces, the middle peak corresponds to structures where aromatic ring is parallel to the surface and the amine group is rotated away from the surface by one carbon bond in the linker, as in structures
(v) and
(viii).
The third peak from the surface represents the most probably configuration for all curvatures. In these structures, the two linker bonds that connect the plane of the aromatic ring to the amine group are both oriented to extend the amine out away from the surface (see structures
in
Figure 8c). The location of this third peak displays the smallest curvature dependency, as can be seen in the relatively small range in the most probable distances listed in
Figure 8b, third column. Although the structures shown in
Figure 8c only include the neutral DA species, this third peak is the only one observed for the positively charged species, DAH
and DOQH
(see the last two rows of
Figure 7, right-most column). This result indicates that, when protonated, the amine group remains fully extended into the solvent for all curvatures, similar to structures
. These structures also aid in the interpretation of the amine distributions for DA and DOQ at the carbon:vacuum interface in
Figure 7 as well. As compared to the same amine distance distributions at the carbon:water interface, the peak locations remain unchanged, but the relative peak heights shift, indicating that configurations with the amine extending away from the surface are significantly less probable in the absence of solvent.
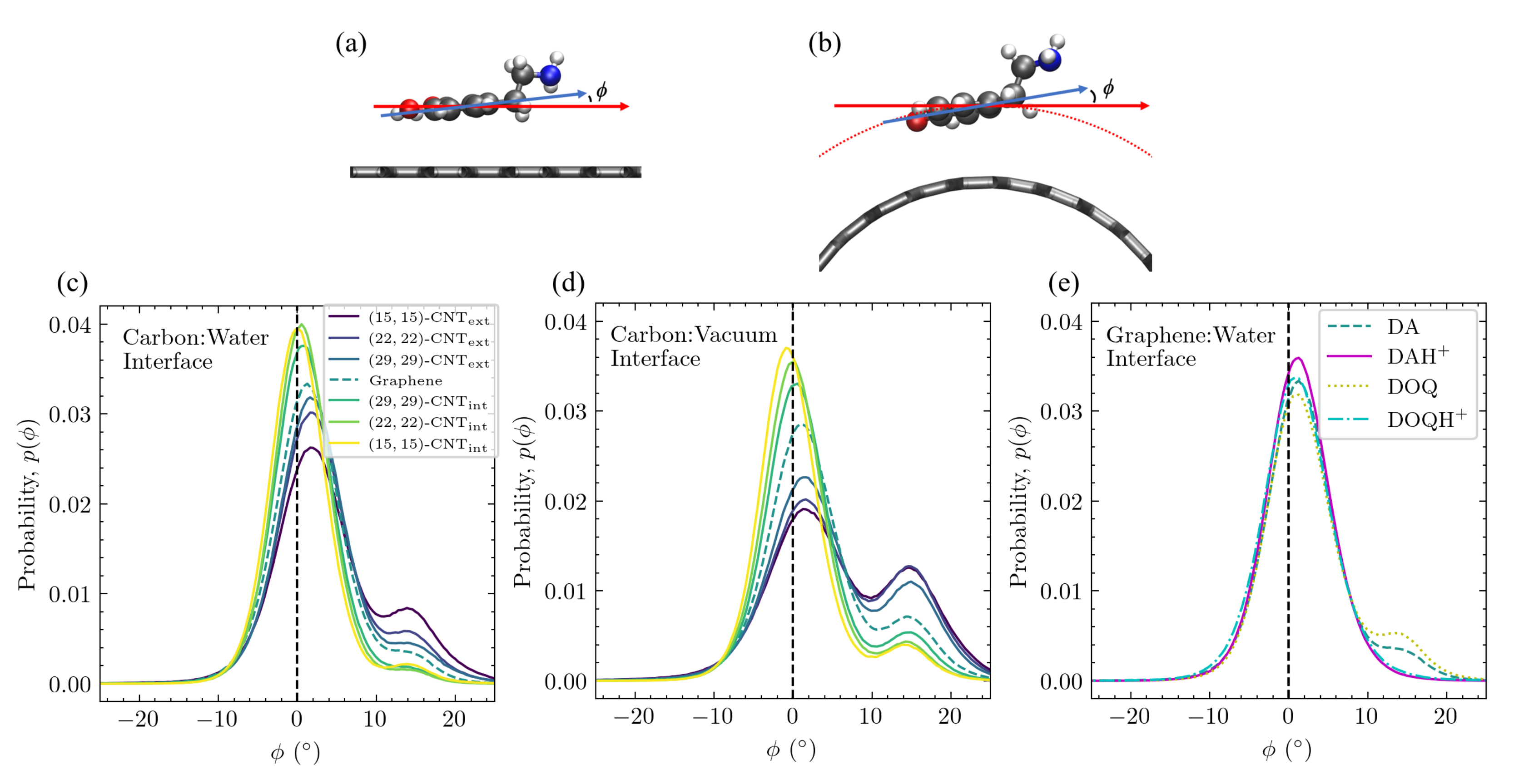
The aromatic ring’s tilt angle above the surface depends on curvature, charge, and solvation. The distributions of the tilt angle between the aromatic ring and the surface are shown in
Figure 9 for DA on differently curved carbon surfaces at both the carbon:water (
Figure 9c) and carbon:vacuum (
Figure 9d) interfaces. In addition, the tilt distributions of DA, DOQ, and their protonated species are shown for flat graphene at the aqueous interface in
Figure 9e.
When adsorbed on all carbon surfaces, DA primarily adopts configurations in which its aromatic ring is parallel to the surface, as seen from the dominant peak, which is close to
in all cases. This configuration maximizes the
–
interactions and is seen in most of the structures shown in
Figure 8c. However, a second, asymmetric peak is observed in a subset of the cases at
and corresponds to the tentlike configurations seen in structures
(i) and
(ii) in
Figure 8c. The relative probability of these two tilt angles clearly depends on the surface curvature—the peak at
is strongest on the most concave surface, whereas the peak at
is strongest on the most convex surface.
The tilt angle distributions also display a clear dependence on charge, as can be seen on solvated flat graphene in
Figure 9e, where there is substantial probability around
for DA and DOQ but no such density for DAH
and DOQH
. From the amine group distributions for these positively charged species in
Figure 7, we know that they adopt configurations in which the amine group stretches out into the bulk water, which precludes the more tilted tentlike structures like
(i) and
(ii) in
Figure 8c.
The tilt angle distribution also depends on solvation. As can be seen in
Figure 9d, the curvature-dependence observed at the carbon:water surface is also present at the carbon:vacuum surface. However, the population of the tilted configuration increases in all cases, which corresponds well to the shift in the amine distance distribution to values that are closer to the surface for the carbon:vacuum surfaces in
Figure 7. Similar results were obtained for DOQ at the carbon:vacuum interface, see
Figure S6a.
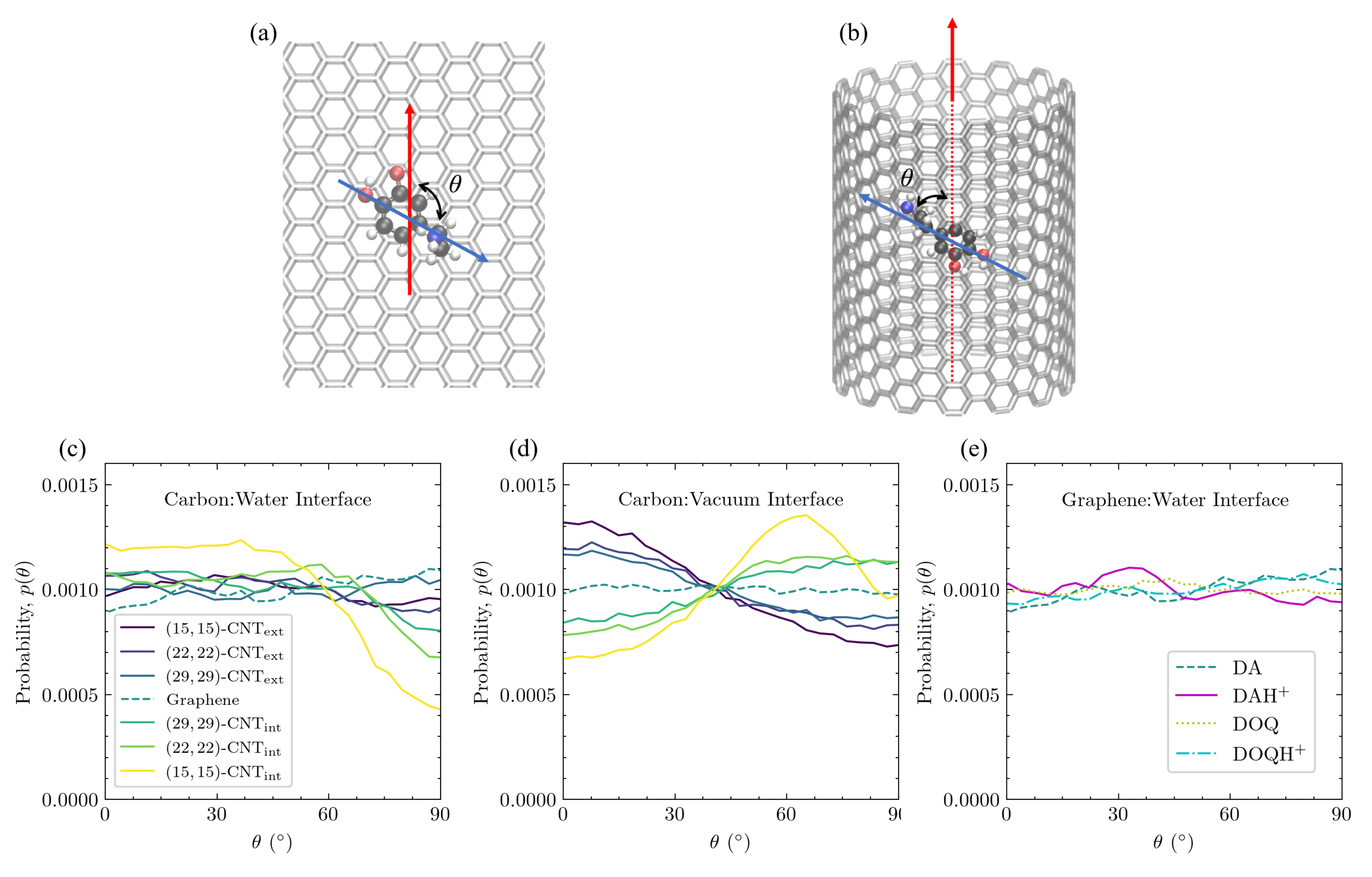
Adsorbate alignment with CNT axis also depends on curvature and solvation. In
Figure 10, the orientational alignment of DA with the axis of the CNT, as defined by
in
Figure 10a,b, is shown on differently curved carbon surfaces at the carbon:water interface (
Figure 10c) and at the carbon:vacuum interface (
Figure 10d). The
distributions of DA, DOQ, and their protonated species are also shown for flat graphene at the aqueous interface in
Figure 10e.
At the carbon:water interface, the
distribution of DA is uniform on flat graphene and on the exterior of the CNTs, as can be seen in
Figure 10c. In addition, charge does not seem to influence this orientation for the solvated flat graphene case shown in
Figure 10e. Even the flat graphene case at the carbon:vacuum interface in
Figure 10d shows no change in the probability with
. This invariance of the probability of a given
orientation on flat graphene is to be expected, given the lack of curvature to break the symmetries present in the flat graphene case as well as the lack of significant lateral distribution patterning in
Figure 4.
In contrast, on the solvated CNT interior in
Figure 10c, there is a marked decrease in the orientational probability as
approaches
, and the effect becomes more dramatic as the concavity increases. These highly curved interior surfaces favor orientations where the longest axis of DA is oriented along the CNT axis (
). This orientational preference is linked to the rightward shift in the aromatic ring’s vertical distribution as the surface changes from flat to concave, as shown in
Figure 7a. In configurations where DA is not aligned with the CNT axis, its interactions with the inward-curving walls will force the center of the ring slightly away from its optimal distance above the surface. The data obtained from ten trajectories show that, on the solvated interior of the
-CNT nanotube, where this orientational preference is strongest, the average vertical distance of the ring’s COM for all configurations in which DA is closely aligned with the CNT axis (
) is
Å, whereas the average vertical distance for the configurations where DA is perpendicularly aligned to the CNT axis (
) is
Å.
The
distribution is entirely different at the carbon:vacuum interface, however. Orientations aligned with the CNT axis are disfavored on the CNT interior, and the most favorable orientation on the CNT interior shifts to ≈65
. At the same time, orientations aligned with the CNT axis are favored on the CNT exterior. Both trends grow stronger with increased curvature, and the same trends were observed for DOQ at the carbon:vacuum interface (
Figure S6b).
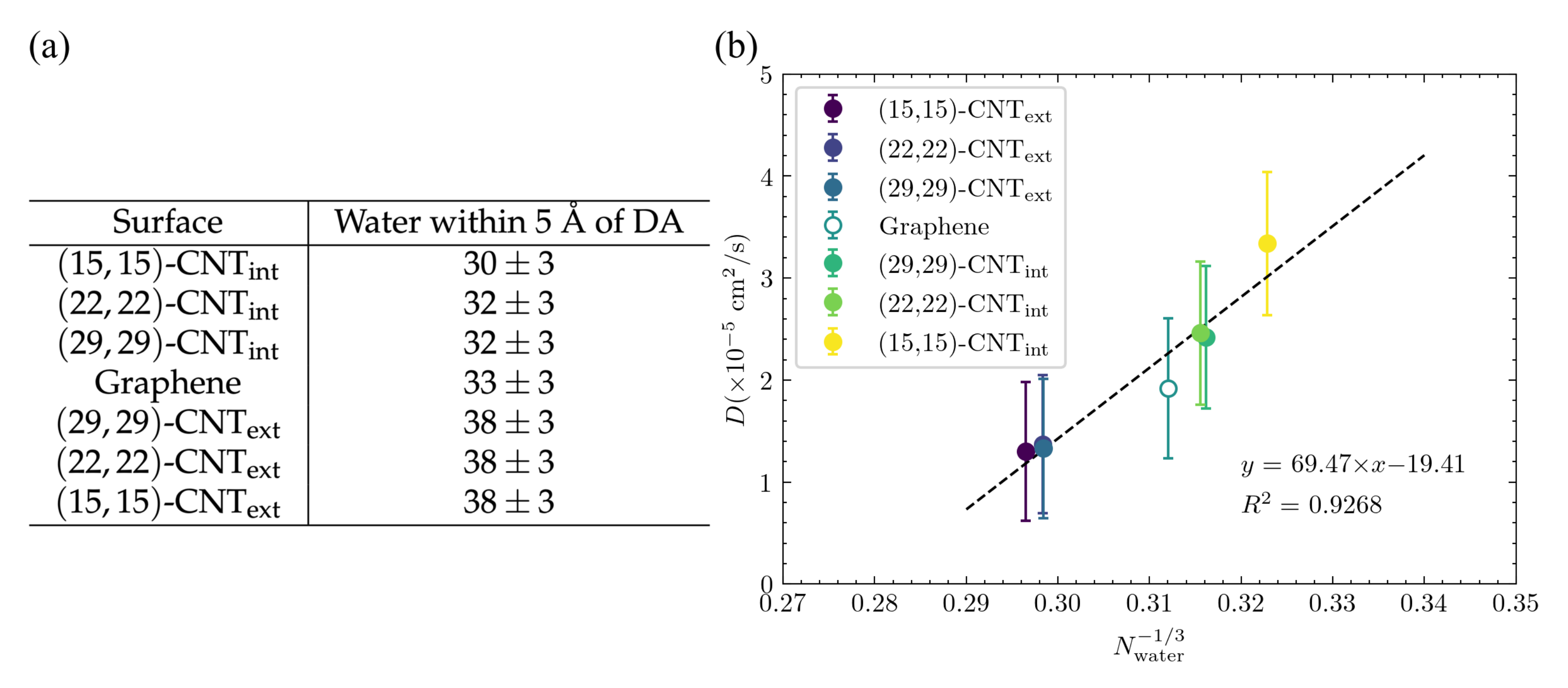
Adsorbate solvation shell depends on curvature and influences . According to the Stokes–Einstein equation, the diffusion constant,
, where
is the effective hydrodynamic radius, which depends on the magnitude of attractions between the diffusing particle and the nearby solvent molecules. In the case of a particle adsorbed to a surface, solvation is necessarily limited by the presence and geometry of that surface. Although the Stokes–Einstein relation cannot be directly applied in that situation, it does provide a way to think about the influence of the degree of solvation on diffusion, as the magnitude of any favorable interactions between the particle and nearby solvent will influence the particle’s effective hydrodynamic radius,
. To investigate this effect, we calculated the number of solvating water molecules within the first water shell around the DA or DOQ atoms for each surface architecture. A distance of 5 Å was chosen as the cutoff of that first water shell based on the distribution shown in
Figure S5. The results are shown in
Figure 11a and display a clear trend from fewest solvating waters on the smallest CNT’s interior to the most solvating waters on the smallest CNT’s exterior—as expected given the geometric constraints of the surface. This trend matches that seen in the diffusion constants on different surface curvatures, as seen in
Figure 3c. To determine how well this solvation effect can explain the trend in diffusivities, we plotted in
Figure 11b the diffusivities from
Figure 3c vs.
, where
is the number of waters within 5 Å of a DA atom, since
, and
is roughly
. The correspondence is quite strong, and this effect is even able to explain the overlapping values seen in the diffusivities even as curvature steadily changes for the CNT exteriors and for the
-CNT
and
-CNT
cases. Since there is a clear geometric trend across these different diameter CNTs, the fact that the number of solvating waters is the same implies that a change in the adsorbate structures, as documented above, must compensate for that change in a way that maintains a similar degree of solvation.
Overall, we find here that the vertical placement of the adsorbate and its moieties above the carbon surface, as well as its tilt angle and alignment with the CNT axis depend in a complex manner on curvature, solvation, and charge. In addition, the degree of DA solvation varies with curvature and can explain much of the trend observed as D varies across curvatures.
3.4. DA Localizes and Diffuses within a CNT Groove
Since a pair of aligned CNTs is the simplest multi-CNT structure expected within CNT-based material, we also investigated how DA behaves on a groove surface, both for the solvated and vacuum cases.
In all the simulations, DA localized to the groove between the two CNTs within 2–8 ns and remained there. Given this strong structural preference, we ran each simulations for at least 5 ns after it found its way to the groove. All results presented in this section were obtained from the portions of the trajectories where DA is within the CNT groove.
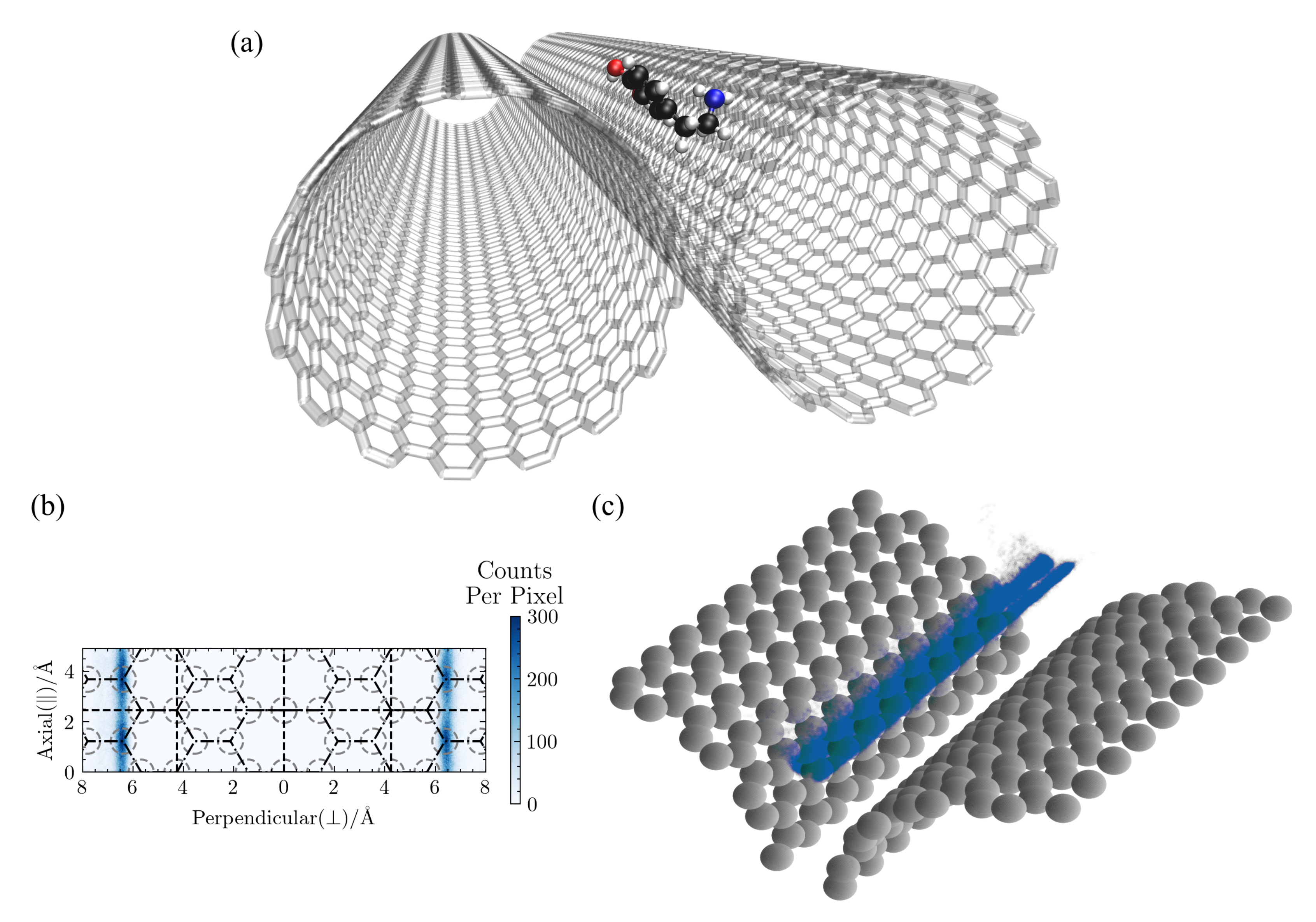
A typical configuration from a groove simulation is shown in
Figure 12a, the lateral distribution of DA is shown in
Figure 12b, and the 3D distribution is shown in
Figure 12c. There is a slight dependence on the underlying hexagonal structure in the lateral density distribution in
Figure 12b, but only the axial direction, as the adsorbate’s location around the circumference is determined by the optimal distance from the other CNT surface, as can be seen in
Figure 12c. Note that any lateral patterning will depend on the degree to which the neighboring CNTs are in register. There is a clear separation in the 3D density plot between configurations with DA adsorbed to one CNT surface vs. the other. Jumps between the two CNT surfaces are rare in the solvated case (
ns
), but were more frequently for DA adsorbed at the carbon:vacuum interface (
ns
). Jump trajectories across both the carbon:water and carbon:vacuum CNT grooves can be seen in
Figure S7.
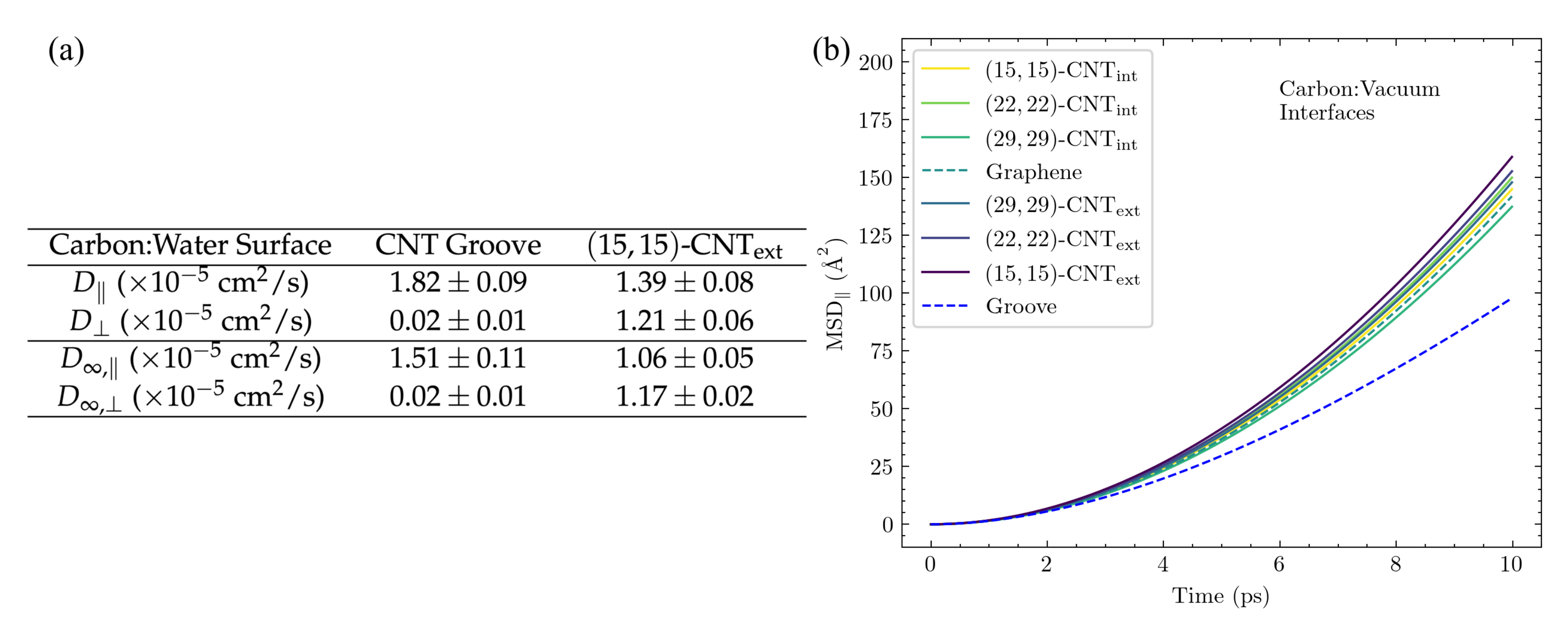
Figure 13a compares
and
for DA in the solvated groove to the same values for DA on the exterior of a single
-CNT, since the groove is constructed of two aligned
-CNTs. Results directly obtained from the 100 Å-length CNT groove system are shown in the top section of the table, while the extrapolation to the infinite CNT groove is shown at the bottom. Importantly, the observed trends hold for both the finite size results and the infinite size extrapolations. As expected,
drops to almost zero when DA remains in the groove. In contrast, DA’s diffusivity along the groove,
, is significantly faster than the corresponding axial diffusivity on the exterior surface of a single CNT. We then calculated
for DA in the groove and found
waters within 5 Å. This value is lower than those reported in
Figure 11a for the other surface structures and explains the faster diffusion within the solvated groove.
Interestingly, this trend is reversed for DA’s diffusivity in the groove at the carbon:vacuum interface.
Figure 13b shows the comparison of the axial MSD of DA in the groove to that of DA on other CNT and graphene surfaces, all at the carbon:vacuum interface. Without solvent, displacement along the groove is reduced as compared to that on any other surface, which can be readily explained by the presence of two variegated surfaces that can impact DA’s inertial motion rather than just one.
The results for the diffusion of all four adsorbate species within the 100 Å CNT groove are shown in
Table 3. The previously observed trends between oxidized and reduced species (oxidized diffuses more rapidly) and between protonated and neutral species (neutral diffuses more rapidly) both hold within the groove architecture.

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



